全身型重症筋無力症患者へのソリリス®投与における質問集(FAQ)
ソリリス®投与に関するよくある質問をまとめました
ソリリス®とは?
- ソリリス®はどんな薬ですか?
-
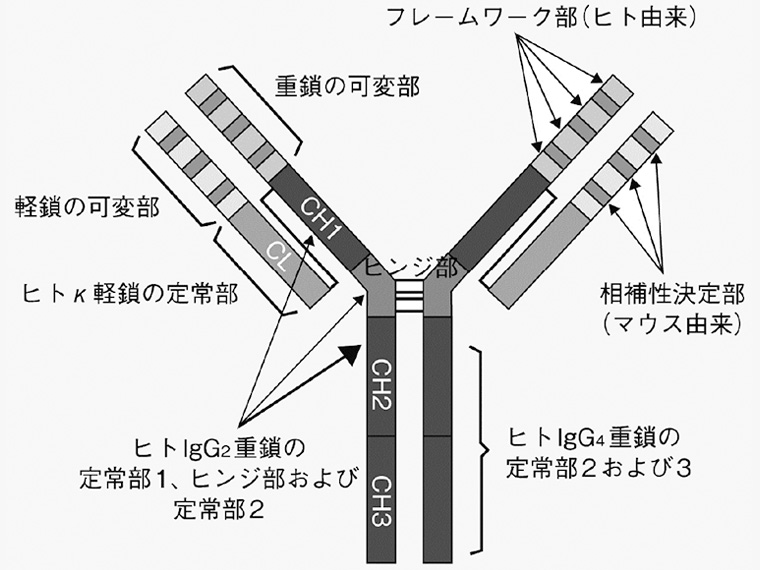
ソリリス®はエクリズマブ(遺伝子組換え)を有効成分とする遺伝子組換えヒト化モノクローナル抗体であり、マウス抗ヒト補体C5α鎖抗体の相補性決定部及びヒトフレームワーク部からなります。
<エクリズマブの基本構造概略図>
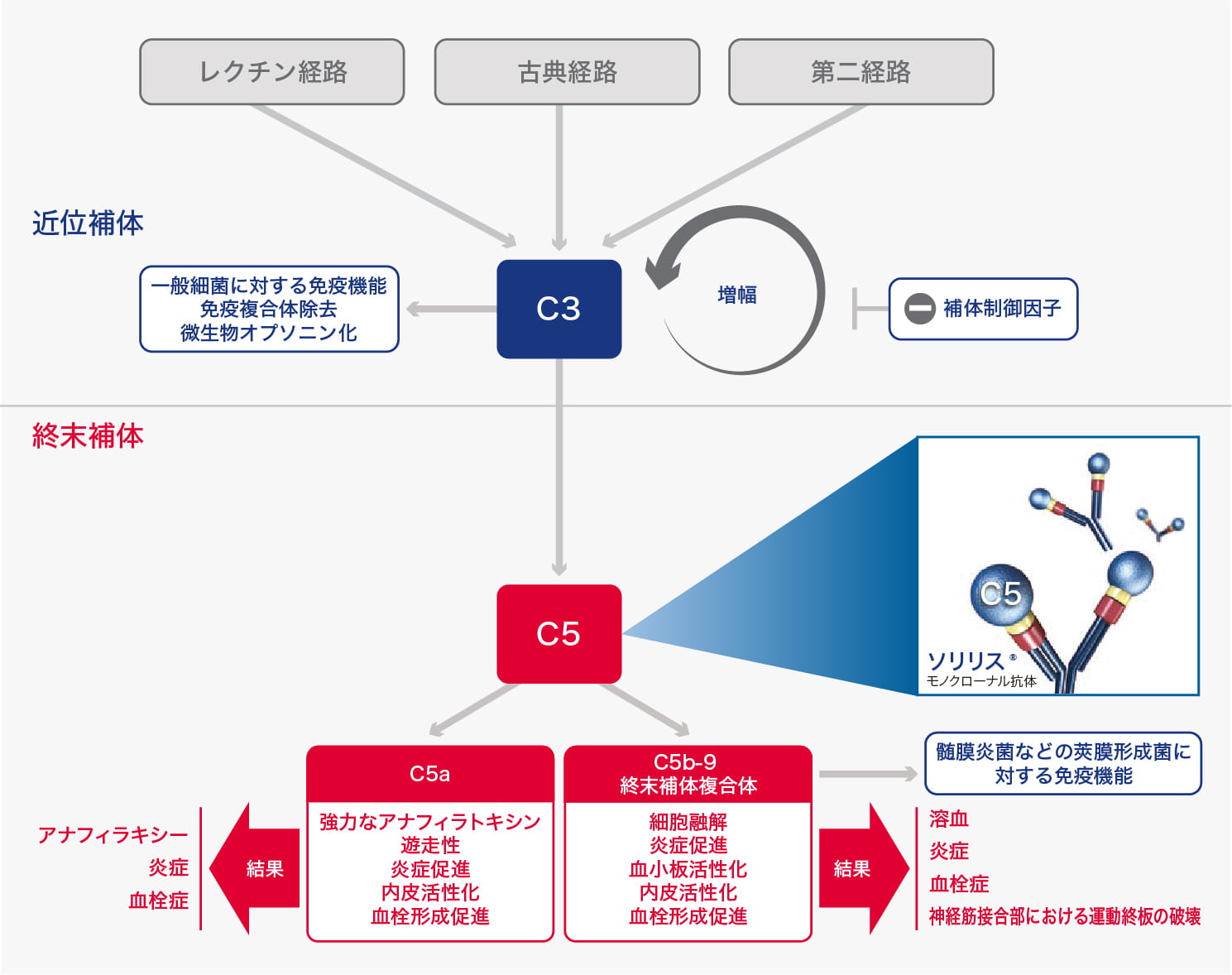
ソリリス®は、補体カスケードにおけるヒトC5に特異的に結合し、その開裂を用量依存的に阻害し、C5b-9からなる膜侵襲複合体(MAC)の形成を抑制すると考えられます1)。
<ソリリス®の作用機序>
引用 1) Rother RP, et al. Nat Biotechnol. 2007; 25(11): 1256-1264.
[利益相反:本論文の著者にはAlexion Pharmaceuticals, Inc.の社員が含まれます]関連する質問
電話でのお問い合わせ
フリーダイヤル 0120-577-657
受付時間 9:00 ~ 17:30(土、日、祝日及び当社休業日を除く)
- 成人のMG患者を対象とした臨床試験について教えてください
-
成人のMGに対するソリリス®の適応は、難治性の全身型MG患者を対象とした海外第Ⅱ相臨床試験C08-0011)、第Ⅲ相国際共同臨床試験(REGAIN試験)及びREGAIN継続試験の3試験結果が、承認時評価資料とされました。ここでは、REGAIN試験及びREGAIN継続試験の結果についてご紹介します。 一部承認外のデータが含まれますが、承認時評価資料ですのでご紹介します。 ●第Ⅲ相国際共同臨床試験(REGAIN試験)2, 3) (無作為化二重盲検プラセボ対照第Ⅲ相並行群間多施設共同試験) 【試験概要】
- ・目的:難治性の全身型MG患者に対するソリリス®の有効性と安全性を評価する。
- ・対象:難治性の全身型MG患者125 例
- ・方法:ソリリス®投与開始の2週以上前に髄膜炎菌に対するワクチンを接種した。ソリリス®900mg又は等量のプラセボを週1回、4回点滴静注し、その1週間後(初回投与から4週間後)からソリリス®1200mg又は等量のプラセボを2週に1回の間隔で点滴静注した。
- ・主要評価項目:26 週におけるMG-ADL総スコアのベースラインからの変化量
- ・副次評価項目:26週におけるQMG総スコアのベースラインからの変化量、26週におけるMGC総スコアのベースラインからの変化量、26週におけるMG-QOL15総スコアのベースラインからの変化量 等
- ・解析計画:MG-ADLについて、Worst-Rank ANCOVA(共分散分析)を行った。また、MG-ADL、QMG、MGC、MG-QOL15の各評価項目について、感度分析の実施を事前に規定した。感度分析では、各来院時のベースラインからの変化量を観察し、投与、来院、投与と来院の交互作用、MGFA分類(クラスⅡa又はⅢa/Ⅳa/Ⅱb又はⅢb/Ⅳb)、総スコアのベースライン値を組み込んだ反復測定モデルを用いて解析した。なお、ANCOVAは、26週の欠測値を補完するlast observation carried forward(LOCF)解析を用いた。一方、反復測定解析においては、欠測値の補完は考慮しなかった。
主要評価項目である「26週におけるMG-ADL総スコアのベースラインからの変化量」のWorst-Rank ANCOVAは、以下の表に示す3種類の解析方法で検討された。
治験実施計画書 海外規制当局からの指摘を
踏まえて改訂した解析方法
(統計解析計画書第3.0版の主要解析)事後解析結果※1
(統計解析計画書第2.0版修正版の主要解析)治験実施計画書 - ①レスキュー治療を受けた患者集団(レスキュー治療実施日までの日数が短い順)
- ②レスキュー治療を必要としなかった患者集団[投与26週のMG-ADL総スコアのベースラインからの変化量(LOCF)に基づく改善が小さい順]
海外規制当局からの指摘を踏まえて改訂した解析方法
(統計解析計画書第3.0版の主要解析)- ①投与26週までに死亡した患者集団(死亡した日までの日数が短い順)
- ②MGクリーゼを発現した患者集団(MGクリーゼ発現までの日数が短い順)
- ③レスキュー治療を受けた患者、又は試験を中止した患者集団[レスキュー治療実施日又は中止日(両方のイベントがある場合には早く発現した方)までの日数が短い順]
- ④レスキュー治療を必要とせず26週間の治験薬投与を完了した患者
事後解析結果※1
(統計解析計画書第2.0版修正版の主要解析)- ①投与26週までに死亡した患者集団(死亡した日までの日数が短い順)
- ②MGクリーゼを発現した患者集団(MGクリーゼ発現までの日数が短い順)
- ③レスキュー治療を受けた患者、又は試験を中止した患者のうちレスキュー治療の実施基準に該当する患者集団[レスキュー治療実施日又は中止日(両方のイベントがある場合には早く発現した方)までの日数が短い順]
- ④レスキュー治療を受けなかった患者、又は試験を中止した患者のうちレスキュー治療の実施基準に該当しなかった患者[投与26週のMG-ADL総スコアのベースラインからの変化量(LOCF)に基づく改善が小さい順]
※1 試験を中止した患者をより適切に取り扱うため、事後解析として新たなWorst-Rank解析を実施した。
【有効性:主要評価項目】
- ・主要評価項目[統計解析計画書第2.0版修正版(事後解析計画)]である26週におけるMG-ADL総スコアのベースラインからの変化量において、ソリリス®群とプラセボ群の間に統計学的に有意な差が認められました(p=0.0160、Worst-Rank ANCOVA※2) 。
- ・主要評価項目(統計解析計画書第1.0版)である26週におけるMG-ADL総スコアのベースラインからの変化量において、ソリリス®群とプラセボ群の間に統計学的に有意な差が認められました(p=0.0089、Worst-Rank ANCOVA※2)。
- ・主要評価項目(統計解析計画書第3.0版)である26週におけるMG-ADL総スコアのベースラインからの変化量において、ソリリス®群とプラセボ群の間に統計学的に有意な差は認められませんでした(p=0.0698、Worst-Rank ANCOVA※2)。
※2 順位を応答変数とした投与群及びMGFA分類(クラスⅡa又はⅢa/Ⅳa/Ⅱb又はⅢb/Ⅳb)を因子、MG-ADL総スコアのベースライン値を共変量とした。
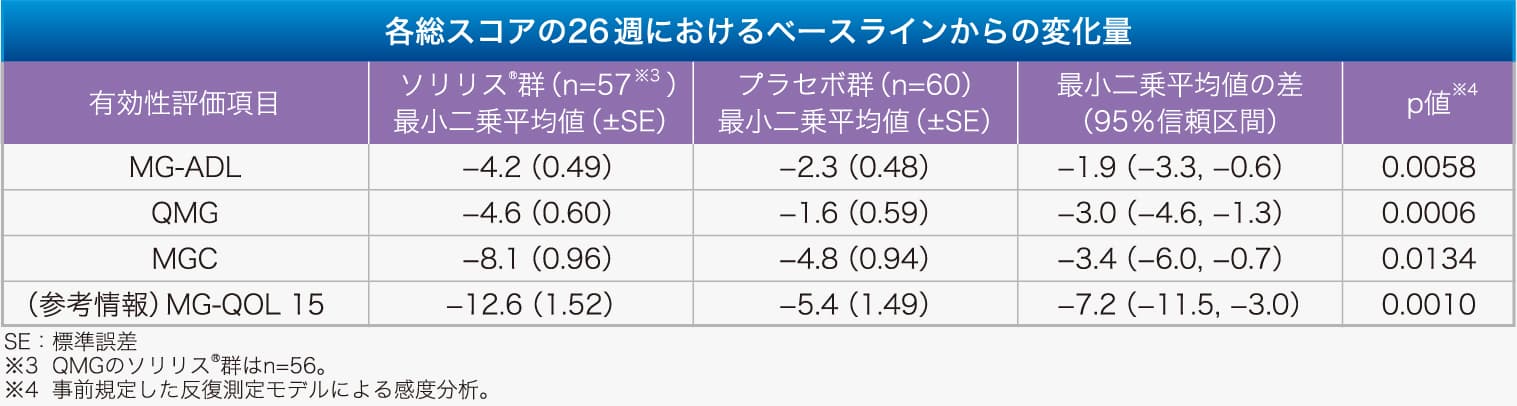
【有効性:主要評価項目(MG-ADL)、副次評価項目(QMG、MGC、参考情報:MG-QOL15)】 各総スコアの26週におけるベースラインからの変化量において、ソリリス®群とプラセボ群の間に統計学的に有意な差が認められました。
【安全性】ソリリス®群の53例(85.5%)及びプラセボ群の56例(88.9%)に有害事象が認められました。 ソリリス®群で認められた主な有害事象は、頭痛10例(16.1%)、上気道感染10例(16.1%)、鼻咽頭炎9例(14.5%)、悪心8例(12.9%)、下痢8例(12.9%)、重症筋無力症6例(9.7%)でした。プラセボ群で認められた主な有害事象は、頭痛12例(19.0%)、上気道感染12例(19.0%)、重症筋無力症11例(17.5%)、鼻咽頭炎10例(15.9%)、悪心9例(14.3%)、下痢8例(12.7%)でした。重篤な有害事象はソリリス®群の9例(14.5%)及びプラセボ群の18例(28.6%)に認められ、重症筋無力症[ソリリス®群:5例(8.1%)、プラセボ群:8例(12.7%)]、発熱[ソリリス®群:2例(3.2%)、プラセボ群:0例]及び上気道感染[ソリリス®群:0例、プラセボ群:2例(3.2%)]等でした。投与中止に至った有害事象はソリリス®群の4例に認められ、重症筋無力症クリーゼ、腸管穿孔、前立腺癌、菌血症でした。試験期間中に死亡した例あるいは注目すべき有害事象(AESI)として定義された髄膜炎菌感染症を発現した例はありませんでした。なお、重症筋無力症クリーゼはソリリス®群で1例に認められました。
●第Ⅲ相国際共同臨床試験(REGAIN継続試験)4) (非盲検多施設共同試験)
【試験概要】
- ・目的:ソリリス®長期投与時の有効性と安全性を評価する。
- ・対象:REGAIN試験を完了した難治性の全身型MG患者117例
- ・方法:REGAIN試験の26週目の来院完了後2週間以内に本試験に移行することとした。REGAIN試験のソリリス®群の患者にはDay1及び2週目にソリリス®1200mgを、1週目と3週目にプラセボを投与し、REGAIN試験のプラセボ群の患者にはDay1及び1〜3週目にソリリス®900mg+プラセボを投与した。REGAIN試験の盲検性を維持するため、4週間は投与及び評価を盲検下で実施した。4週目から試験終了時まで、ソリリス®1200mgを2週間に1回投与した。
- ・主要評価項目:MG-ADL総スコアのソリリス®/ソリリス®群及びプラセボ/ソリリス®群のベースラインからの変化量
- ・副次評価項目:QMG総スコアのベースラインからの変化量 等
- ・解析計画:2016 年9 月21 日をデータカットオフ日とし、中間解析を実施した。有効性は、継続試験のFAS(REGAIN継続試験でソリリス®を1回以上投与された患者で、治験薬投与後に有効性評価を1回以上受けた全患者)を用い、①REGAIN継続試験ベースライン(Day1の評価)、②REGAIN試験ベースライン[REGAIN試験の有効性の解析で用いたベースライン(Day1の評価)]、以上2つのベースラインを用いて解析した。安全性については、本試験でソリリス®を1回以上投与された患者を用いて解析した。有効性評価項目に関しては、総スコアのベースライン値及び来院を組み込んだ反復測定モデルを用いて比較検討した。
【有効性:MG-ADL総スコア(主要評価項目)】
- ・52週間のソリリス®投与期間を通じて、REGAIN試験の26週と同程度のMG-ADL総スコアのベースラインからの改善が認められました。
- ・REGAIN試験でプラセボを投与され、本試験でソリリス®を投与された患者群(プラセボ/ソリリス®群)において、MG-ADL総スコアに臨床的に意味のある改善※5が認められました。
- ・プラセボ/ソリリス®群では、MG-ADL総スコアのREGAIN継続試験ベースラインからの改善が1週目の早期から認められ[平均値(95%信頼区間) -1.6(-2.28, -0.89)、p<0.0001、反復測定モデル]、治療効果は52週[-2.7(-3.73, -1.63)、p<0.0001、反復測定モデル]まで持続しました。
※5 MG-ADL総スコアのベースラインから3ポイント以上の低下を臨床的に意味のある改善とした。
【有効性:QMG総スコア(副次評価項目)】
- ・52週間のソリリス®投与期間を通じて、REGAIN試験の26週と同程度のQMG総スコアのベースラインからの改善が認められました。
- ・プラセボ/ソリリス®群では、REGAIN試験のソリリス®群と同様に、QMG総スコアに臨床的に意味のある改善※6が認められました。
- ・プラセボ/ソリリス®群では、QMG総スコアのREGAIN継続試験ベースラインからの改善が1 週目の早期から認められ[平均値(95%信頼区間) -2.3(-3.31, -1.37)、p<0.0001、反復測定モデル]、治療効果は52週[-4.6(-6.06, -3.12)、p<0.0001、反復測定モデル]まで持続しました。
※6 QMG総スコアのベースラインから5ポイント以上の低下を臨床的に意味のある改善とした。
【安全性】 REGAIN継続試験に移行した患者のうち、ソリリス®/ソリリス®群の52例(92.9%)及びプラセボ/ソリリス®群の55例(90.2%)に有害事象が認められました。主な有害事象は、ソリリス®/ソリリス®群では鼻咽頭炎15例(26.8%)、頭痛11例(19.6%)、関節痛10例(17.9%)、下痢9例(16.1%)、重症筋無力症9例(16.1%)、上気道感染8例(14.3%)、プラセボ/ソリリス®群では、頭痛20例(32.8%)、鼻咽頭炎13例(21.3%)、下痢8例(13.1%)、重症筋無力症8例(13.1%)、悪心8例(13.1%)、四肢痛7例(11.5%)でした。 重篤な有害事象はソリリス®/ソリリス®群の17例(30.4%)及びプラセボ/ソリリス®群の18例(29.5%)に認められ、5%以上に認められた重篤な有害事象はいずれの群も重症筋無力症でした。投与中止に至った有害事象はプラセボ/ソリリス®群及びソリリス®/ソリリス®群の各1例に認められ、いずれも重症筋無力症でした。有害事象による死亡はソリリス®/ソリリス®群で1例(サイトメガロウイルス関連血球貪食性リンパ組織球症)に認められました。
臨床試験の詳細をまとめたパンフレットがあります。ご参照ください。
引用 1)社内資料:海外第Ⅱ相臨床試験(C08-001)(承認時評価資料) 2)社内資料:第Ⅲ相プラセボ対照二重盲検比較臨床試験(ECU-MG-301)(承認時評価資料) 3)Howard JF Jr, et al. Lancet Neurol 2017; 16: 976-986. [利益相反:本試験はAlexion Pharmaceuticals, Inc.の支援のもと実施されました] 4)社内資料:第Ⅲ相ECU-MG-301継続試験(ECU-MG-302)(承認時評価資料)
- 4. 効能又は効果(抜粋)
全身型重症筋無力症(免疫グロブリン大量静注療法又は血液浄化療法による症状の管理が困難な場合に限る)
関連する質問
▶ ソリリス®はどんな薬ですか? ▶ 小児のMG患者を対象とした臨床試験について教えてください ▶ MG患者を対象とした臨床効果はいつ頃からみられますか? ▶ 成人のMG患者を対象とした臨床試験ではどのような副作用が認められましたか?電話でのお問い合わせ
フリーダイヤル 0120-577-657
受付時間 9:00 ~ 17:30(土、日、祝日及び当社休業日を除く)
- 小児のMG患者を対象とした臨床試験について教えてください
-
小児のMGに対するソリリス®の用法及び用量の追加は、難治性の全身型MG患者を対象とした第Ⅲ相国際共同臨床試験(ECU-MG-303試験)の結果が、承認時評価資料とされました。 一部承認外のデータが含まれますが、承認時評価資料ですのでご紹介します。 ●第Ⅲ相国際共同臨床試験(ECU-MG-303試験)1) (非盲検単群多施設共同試験) 【試験概要】
- ・目的:難治性の全身型MG小児患者に対するソリリス®の有効性、安全性、薬物動態及び薬力学を評価する。
- ・対象:難治性の全身型MG小児患者11例(6歳以上18歳未満、ただし組み入れられた患者は12歳以上であった)
- ・方法:ソリリス®600~900mgを週1回で計1~4回、その1週間後から300~1200mgを2週に1回点滴静注した。
なお、IVIg※2の維持療法を受けながら本試験に参加する患者には、下表に従いソリリス®の補充投与を行った。体重※1 導入期 維持期 体重※140kg以上 導入期1回900mgを週1回で計4回 維持期初回投与4週間後から1回1200mgを2週に1回 体重※130kg以上40kg未満 導入期1回600mgを週1回で計2回 維持期初回投与2週間後から1回900mgを2週に1回 体重※120kg以上30kg未満 導入期1回600mgを週1回で計2回 維持期初回投与2週間後から1回600mgを2週に1回 体重※110kg以上20kg未満 導入期1回600mgを週1回で計1回 維持期初回投与1週間後から1回300mgを2週に1回 体重※1 導入期 維持期 補充投与量 総投与量 補充投与量 総投与量 体重※140kg以上 導入期 補充投与量600mg 導入期 総投与量1500mg 維持期 補充投与量600mg 維持期 総投与量1800mg 体重※130kg以上40kg未満 導入期 補充投与量300mg 導入期 総投与量900mg 維持期 補充投与量600mg 維持期 総投与量1500mg 体重※120kg以上30kg未満 導入期 補充投与量300mg 導入期 総投与量900mg 維持期 補充投与量300mg 維持期 総投与量900mg 体重※110kg以上20kg未満 導入期 補充投与量300mg 導入期 総投与量900mg 維持期 補充投与量300mg 維持期 総投与量600mg ※1 各投与日の来院時に測定された体重に基づく。 ※2 本邦ではIVIgには小児におけるMGの適応はない。
投与期間は、主要評価投与期が26週間、継続投与期が最長208週間(継続中)とした。 - ・主要評価項目:QMG総スコアのベースラインからの変化量(主要解析は12週時)
- ・解析計画:中間解析として、主要評価投与期での有効性の主要評価項目及び副次評価項目の解析を、ソリリス®を1回以上投与されたすべての患者(FAS)のうち年長の小児患者(12歳以上18歳未満)のみを含むmodified FAS(mFAS)に関して実施した。主要評価項目であるQMG総スコアのベースラインからの変化量は、ベースライン時点のQMG総スコア及び来院を共変量とした反復測定混合効果モデル(MMRM)に基づく制限付き最尤法により、最小二乗平均値が0に等しいかどうかを検定した(主要解析は12週時)。欠測値は補完しなかった。
2022年6月1日を52週時のデータカットオフ日として、長期の有効性及び安全性を評価した。
【有効性:主要評価項目】
- ・QMG総スコアのベースラインからの変化量の最小二乗平均値は、主要解析時点である12週時に-5.2[p=0.0009、ベースライン時点のQMG総スコア及び来院を共変量としたMMRMに基づく制限付き最尤法(最小二乗平均値が0に等しいかを検定)]でした。

SE:標準誤差 *p=0.0009、ベースライン時点のQMG総スコア及び来院を共変量としたMMRMに基づく制限付き最尤法(最小二乗平均値が0 に等しいかを検定)
【安全性】52週データカットオフ日までに、11例全例(100%)に有害事象が発現しました。主な有害事象(20%以上の発現割合)は、発熱、頭痛及び上咽頭炎各4例(36.4%)、腹痛及び四肢痛各3例(27.3%)でした。 重篤な有害事象は3例(27.3%)に認められ、重症筋無力症クリーゼ1例、重症筋無力症及び発熱(重複)1例、扁桃周囲膿瘍1例でした。このうち、扁桃周囲膿瘍は、治験責任医師によりソリリス®との関連性が否定できないと判断されました。 死亡及び投与中止に至った有害事象は認められませんでした。 注目すべき有害事象※3は4例に認められましたが(扁桃周囲膿瘍1例、そう痒性皮疹1例、発疹1例、湿疹・発疹・蕁麻疹1例)、注目すべき有害事象※3として定義された髄膜炎菌感染、アスペルギルス感染及び敗血症は認められませんでした。
※3 注目すべき有害事象:髄膜炎菌感染、アスペルギルス感染、敗血症、その他の重篤な感染及び注入に伴う反応。
臨床試験の詳細をまとめたパンフレットがあります。ご参照ください。
引用 1)社内資料:第Ⅲ相ECU-MG-303試験(承認時評価資料)
- 4. 効能又は効果(抜粋)
全身型重症筋無力症(免疫グロブリン大量静注療法又は血液浄化療法による症状の管理が困難な場合に限る)
関連する質問
▶ ソリリス®はどんな薬ですか? ▶ 成人のMG患者を対象とした臨床試験について教えてください ▶ MG患者を対象とした臨床効果はいつ頃からみられますか? ▶ 成人のMG患者を対象とした臨床試験ではどのような副作用が認められましたか?電話でのお問い合わせ
フリーダイヤル 0120-577-657
受付時間 9:00 ~ 17:30(土、日、祝日及び当社休業日を除く)

- MG患者を対象とした臨床効果はいつ頃からみられますか?
-
ソリリス®の全身型MG患者を対象とした臨床試験では、ほとんどの治療反応例で投与開始後12週までに症状の改善が得られていました。全身型MG患者で他の免疫抑制剤を併用している患者さんにおいては、髄膜炎菌感染症のリスクが高い可能性があることから、リスクベネフィットを考慮し、投与開始後12 週までに症状の改善が認められない患者さんでは、ソリリス®の投与中止を検討してください。
関連する質問
電話でのお問い合わせ
フリーダイヤル 0120-577-657
受付時間 9:00 ~ 17:30(土、日、祝日及び当社休業日を除く)